
The U.S. Food and Drug Administration doesn’t wait for dangerous drugs to reach pharmacy shelves before acting. Instead, it stops them at the border-no inspection needed. Since September 2025, more than $1.8 billion worth of weight-loss drug ingredients have been blocked at U.S. ports of entry. Not because they were found to be harmful on the spot, but because the manufacturers behind them weren’t on the FDA’s approved list. This isn’t a random crackdown. It’s a system called Import Alerts, and it’s changing how the entire global pharmaceutical supply chain operates.
What Is an FDA Import Alert?
An FDA Import Alert is a public notice that tells customs officials to automatically hold shipments from specific manufacturers or countries without needing to physically test every package. Think of it like a blacklist-but with color codes. There are three levels: Green, Yellow, and Red. Only Green-listed manufacturers get automatic clearance. Everyone else gets detained. This system isn’t new. It’s been around since 1995, but it’s never been this aggressive. Before 2025, most import alerts targeted things like contaminated supplements or poorly labeled medical devices. But in September 2025, the FDA dropped Import Alert 66-80, targeting active pharmaceutical ingredients (APIs) for GLP-1 drugs like semaglutide and tirzepatide. These are the raw chemicals used in popular weight-loss and diabetes medications. The move was a direct response to a flood of unapproved, untested versions of these drugs flooding into the U.S. through third-party suppliers. The FDA didn’t just say, “Don’t import these.” They built a digital wall. Every shipment entering the U.S. is scanned through the Automated Commercial Environment (ACE) system. If the manufacturer isn’t on the Green List, the shipment gets flagged within five business days. No paperwork, no lab test, no second chance. Just detention.How the Green List Works
The Green List is the only way out of detention. It’s not a reward-it’s a requirement. To get on it, manufacturers must prove they meet U.S. quality standards, not just their own country’s. That means more than ISO certifications or local inspections. They need FDA-recognized third-party audits, stability testing across three temperature zones (2-8°C, 25°C, and 40°C), and full traceability of every raw material back to its source-even Tier 3 suppliers. The process takes months. On average, it takes 117 hours of preparation, $45,000 to $68,000 in audit costs, and at least three consecutive compliant shipments before approval. And even then, it’s not guaranteed. The FDA looks at everything: how the facility handles water for injection, whether the lab technicians are trained on U.S. Pharmacopeia standards, whether batch records are digitally signed and tamper-proof. Companies that get it right see results. Pfizer, after installing MediLedger blockchain tracking across 17 API suppliers, hit a 99.8% Green List clearance rate. That’s not luck. That’s infrastructure. But for most small and mid-sized manufacturers-especially in India, where 82% of the affected facilities are located-the cost and complexity are overwhelming.Who Gets Hit the Hardest?
The numbers don’t lie. Of the 89 manufacturers caught under the GLP-1 Import Alert, 73 are in India. Nine are in China. Seven are in Europe. That’s not because Indian manufacturers are worse-they’re just the biggest suppliers. Many of them built their businesses on producing generic APIs cheaply for global markets. Now, they’re being asked to upgrade to U.S. standards overnight. One manufacturer in Hyderabad posted on Reddit: “We had ISO 9001, we had clean inspection reports from our local regulator. But the FDA didn’t recognize our auditor. Our $1.2 million shipment was refused. In 72 hours.” It’s not just about safety. It’s about paperwork. The most common reasons for refusal? Improper Certificate of Analysis (41.7% of cases), missing master production records (33.8%), and no traceability of raw materials (28.5%). These aren’t contamination issues-they’re documentation failures. But under the Import Alert system, documentation failures are treated as safety failures.
The Cost of Getting It Wrong
Getting caught on the wrong side of an Import Alert isn’t just embarrassing-it’s financially devastating. Once a shipment is detained, the importer has 90 days to either export it or destroy it. If they don’t, the U.S. Customs and Border Protection (CBP) can seize the goods and charge liquidated damages up to three times the commercial value. For a $900,000 shipment, that’s $2.7 million in penalties. Some companies are trying to outsmart the system. ProPublica found that some firms are paying brokers to falsify export documents to make it look like the drugs are being sent to Canada or Mexico-when they’re actually being rerouted back into the U.S. The FDA issued a warning letter in October 2025 to a Singapore-based intermediary caught doing exactly that. The ripple effects are huge. Viatris reported a $417 million revenue hit in Q3 2025. Pharmacy benefit managers say prices for compounded GLP-1 formulations have jumped 14.3% since October. And with 28,500 jobs at risk in India alone, the human cost is real.Why This System Exists
The FDA doesn’t do this for fun. In September 2025, a technical review from the American Association of Pharmaceutical Scientists found that 68.4% of refused GLP-1 shipments contained impurities exceeding international safety limits. Some batches had heavy metals. Others had incorrect potency-doses too high or too low. One batch had a contaminant linked to kidney damage. But here’s the twist: 22.1% of refused shipments actually met pharmacopeial standards. They were safe. But they didn’t have the right paperwork. That’s the gray area. Critics argue the system is too blunt. Dr. Rachel Sherman, former FDA deputy commissioner, warned that “automatic detention creates artificial scarcity” and pushes patients toward even riskier gray-market suppliers. The FDA’s response? The Green List isn’t meant to be a punishment. It’s meant to be a roadmap. And they’re making it easier. In November 2025, they cut approval time from 90 days to 45 for companies using FDA-recognized auditors. They also launched the API Transparency Portal, where manufacturers can check their status in real time.What Happens Next?
This isn’t just about weight-loss drugs. FDA Commissioner Dr. Robert Califf said in November 2025 that the same framework will be extended to all high-risk biologics starting in Q1 2026-starting with monoclonal antibodies. That means cancer treatments, autoimmune therapies, and other complex biologics will soon face the same scrutiny. China’s NMPA has already announced it will require FDA-equivalent certifications for all API exports to the U.S. starting January 1, 2026. The European Medicines Agency is moving toward similar automated screening by mid-2026. This is becoming the global standard. Manufacturers who don’t adapt won’t just lose access to the U.S. market-they’ll lose access to the global market. Buyers everywhere are starting to ask: “Are you on the FDA Green List?” If you’re not, you’re seen as high-risk.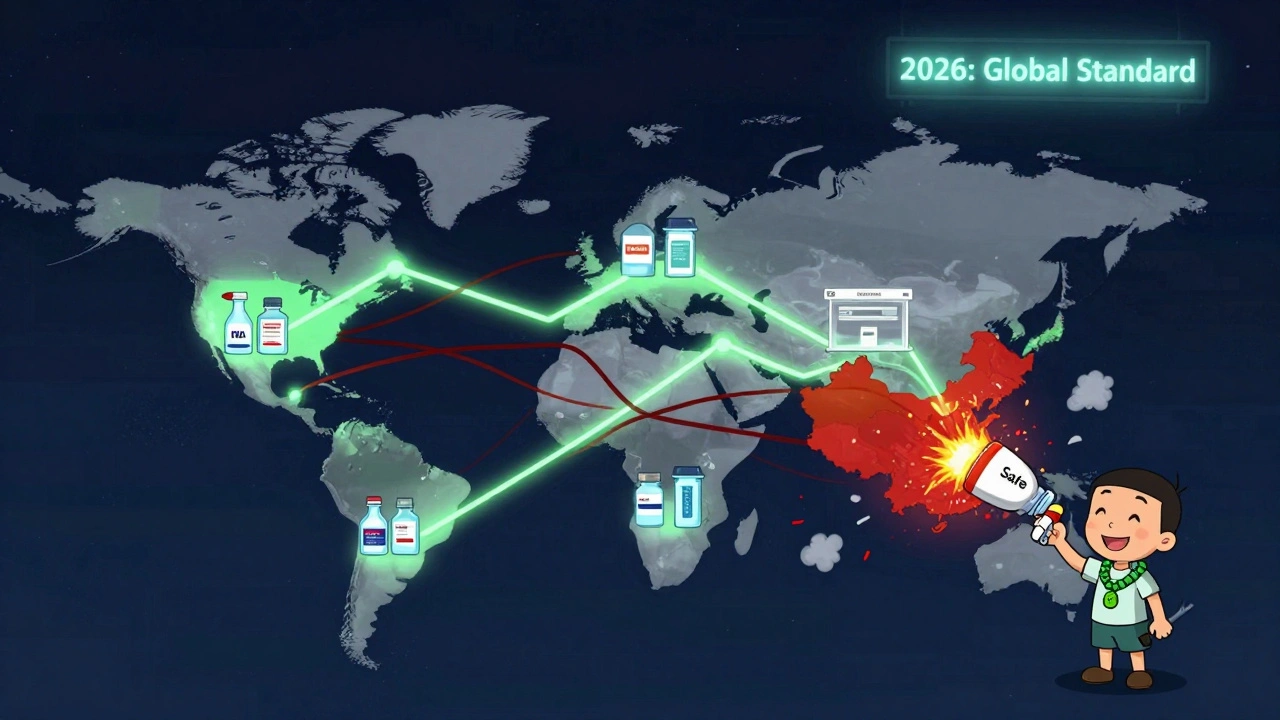
What Manufacturers Can Do
If you’re a manufacturer outside the U.S., here’s what you need to do now:- Check the FDA’s Import Alert list. Know if your facility is flagged.
- Find an FDA-recognized third-party auditor. Not just any auditor. One approved by the FDA.
- Map your entire supply chain. Down to Tier 3. You need to prove where every ingredient came from.
- Upgrade your documentation. Certificates of Analysis must follow FDA format. No exceptions.
- Invest in traceability. Blockchain systems like MediLedger aren’t optional anymore-they’re baseline.
- Start the Green List application early. The process takes months. Don’t wait until your shipment is detained.
What Importers Should Know
If you’re importing drugs into the U.S., you’re responsible for compliance-even if you didn’t make the product. You can’t blame the manufacturer. The FDA holds you accountable.- Never assume a manufacturer is safe because they’ve shipped to you before.
- Verify their status on the FDA’s Green List before placing any order.
- Include compliance clauses in contracts. Make them responsible for FDA violations.
- Don’t rely on “standard” certifications like ISO or CE. They mean nothing under Import Alerts.
- Work with a customs broker who understands PREDICT and ACE systems. Most don’t.
Is This Fair?
Some say it’s protectionism. Others say it’s public health. The truth is somewhere in between. The system isn’t perfect. It’s blunt. It’s expensive. It’s disruptive. But when a patient gets a counterfeit version of semaglutide with no active ingredient-or worse, a toxic impurity-the consequences are irreversible. The FDA’s goal isn’t to punish manufacturers. It’s to protect patients. And right now, the only way to do that at scale is to stop shipments before they even reach the warehouse. The global drug supply chain is changing. The old way-cheap, fast, and loosely regulated-is over. The new way is transparent, documented, and certified. The companies that adapt will survive. The ones that don’t? They’ll be left behind.What happens if a drug shipment is detained under an FDA Import Alert?
The shipment is held at the U.S. port of entry and cannot enter the country. The importer has 90 days to either export the goods back out of the U.S. or destroy them under FDA supervision. If neither is done, Customs and Border Protection can seize the shipment and impose penalties up to three times the commercial value of the goods. Liquidated damages can exceed $2 million for a single shipment.
How can a manufacturer get removed from an FDA Import Alert?
To get removed, a manufacturer must complete four steps: pass a full FDA inspection (minimum 5 days), submit a root cause analysis with a corrective action plan (CAPA), ship three consecutive batches that pass FDA review, and submit an executive certification of compliance. The average time to removal is 11.7 months. Successful petitions often include video evidence of corrected processes, not just documents.
What is the FDA Green List, and how do you get on it?
The FDA Green List is a registry of manufacturers whose products are exempt from automatic detention under Import Alerts. To qualify, manufacturers must undergo an FDA-recognized third-party audit, provide stability testing data across three temperature conditions, map their supply chain to Tier 3 suppliers, and submit detailed batch documentation. The process takes 3-6 months and costs $45,000-$68,000 in audit fees alone.
Why are so many Indian manufacturers affected by the GLP-1 Import Alert?
India is the world’s largest producer of generic active pharmaceutical ingredients (APIs), including those used in GLP-1 drugs. About 82% of the 89 facilities targeted by the September 2025 Import Alert are in India. Many of these manufacturers built their business on low-cost, high-volume production without full U.S. compliance infrastructure. The new rules require costly upgrades in documentation, testing, and traceability that many haven’t yet implemented.
Do FDA Import Alerts apply to over-the-counter drugs and supplements?
Yes. While the recent GLP-1 alert focuses on prescription APIs, Import Alerts have long been used for supplements, OTC drugs, and medical devices. The FDA has over 238 active Import Alerts across all product categories. Any product that fails to meet U.S. safety, labeling, or manufacturing standards can be subject to automatic detention, regardless of whether it’s prescription or over-the-counter.
Will other countries adopt similar import alert systems?
Yes. The European Medicines Agency (EMA) announced in November 2025 that it will adopt similar automated screening protocols for high-risk APIs by Q2 2026. China’s NMPA will require FDA-equivalent facility certifications for all API exporters to the U.S. starting January 1, 2026. The FDA’s system is becoming the global benchmark for drug import safety.
Comments (15)
-
Rupa DasGupta December 5, 2025This is why my cousin’s factory in Hyderabad got crushed. $1.2M gone in 72 hours. ISO 9001? Doesn’t mean squat to the FDA. They don’t care about your hard work. Just your paperwork. 😭

-
Krishan Patel December 5, 2025Let’s be real. This isn’t about safety. It’s about control. The FDA doesn’t want competition. They want monopolies. And they’re using ‘public health’ as a cover to crush small manufacturers who can’t afford their extortionate audit fees. This is capitalism with a side of tyranny.

-
Marvin Gordon December 5, 2025I work in pharma logistics. This system is brutal but necessary. I’ve seen what happens when unregulated APIs slip through. People get sick. Not because of the drug-but because of the shit mixed in with it. The Green List isn’t perfect, but it’s the best damn firewall we’ve got.

-
ashlie perry December 7, 2025They’re using this to hide the fact that Big Pharma is scared of generics. The FDA’s been pressured by Pfizer and Novo Nordisk to kill the competition. The ‘contaminants’? Mostly made up. The real danger is losing your cheap insulin. They’re killing access to medicine under the guise of safety.

-
Stephanie Fiero December 7, 2025If you’re a small manufacturer reading this: stop panicking. Start with the API Transparency Portal. Get your auditor lined up. Map your supply chain like your life depends on it-because it does. You got this. One step at a time. 💪

-
Michael Dioso December 8, 2025Oh wow. So now we’re punishing India because they’re good at making cheap drugs? Next they’ll ban rice because China grows too much. This isn’t regulation. It’s economic warfare dressed up in lab coats. The FDA’s become a trade weapon.

-
sean whitfield December 9, 2025So let me get this straight. You’re telling me a $900k shipment gets destroyed because someone forgot to sign a PDF? Meanwhile, Big Pharma bribes inspectors and ships pills with radioactive isotopes and nobody bats an eye? This isn’t safety. It’s theater.

-
Carole Nkosi December 11, 2025The irony? The same companies that scream about free markets are now demanding the government shut down competitors. Capitalism is dead. What we have now is regulatory feudalism. And the peasants? They’re the ones in Hyderabad and Chennai.

-
Stephanie Bodde December 11, 2025To all the small labs: you’re not alone. I’ve been there. Took us 14 months. But we got on the Green List. It’s not easy. But it’s possible. Reach out. We’ll help. You’re not failing. The system’s just rigged. 🤝

-
Philip Kristy Wijaya December 13, 2025The notion that regulatory compliance constitutes an undue burden is a fallacy rooted in the infantile presumption that market access is a right rather than a privilege contingent upon demonstrable adherence to internationally recognized standards of public health integrity

-
Jennifer Patrician December 15, 2025Did you know the FDA’s database was hacked in 2024? Some manufacturers got moved to Red List overnight because of fake audit reports. Who’s to say this isn’t all just a cover for corporate espionage? The real danger isn’t bad drugs-it’s the system itself.

-
Manish Shankar December 15, 2025As someone who has worked with Indian API manufacturers for over a decade, I can confirm: the majority are not negligent. They lack resources, not intent. The FDA’s approach, while technically sound, ignores the economic reality of global supply chains. A more collaborative path-training, subsidies, phased compliance-would yield better long-term outcomes.

-
luke newton December 17, 2025Let’s not pretend this is about patients. It’s about profits. The same CEOs who make billions off semaglutide are the ones lobbying for these rules. They don’t want cheaper alternatives. They want you dependent on their $1,000/month pills. The FDA? Just their enforcer.

-
Jimmy Jude December 19, 2025The FDA is the new IRS. Only instead of taxes, they tax your entire business model with paperwork. And if you can’t afford the bribe-sorry, ‘audit fee’-you’re out. Welcome to the American Dream™. #RegulatoryCapture

-
Lynette Myles December 20, 2025Green List isn’t a reward. It’s a gate. And the gatekeepers are making sure only the rich get through.
